
中国医科大借iMeta影响因子跃升至33.2(中科院1区)东风,凭多组学联合生信分析成果登刊
2025年6月18日,科睿唯安正式发布了2024年期刊的最新影响因子(发送关键词 “2024JIF” 至 “生信学术纵览”,即可下载完整版最新影响因子 Excel 表格!),这一消息迅速在学术界引发广泛关注。影响因子作为衡量学术期刊影响力的重要指标之一,其每一次变动都如同在学术领域投入一颗石子,激起层层涟漪。众多国际知名期刊的影响因子有了显著变化,如肿瘤神刊 CA: A Cancer Journal for Clinicians 再度暴跌,四大医学期刊继续全线下跌;而 Science 则逆势上涨。在这之中,值得一提的是,国产期刊 iMeta 表现十分亮眼,其影响因子从 2023 年的 23.8 暴涨 40%,一举到达 33.2 的高分,成绩斐然。这一变化不仅体现了 iMeta 期刊影响力的大幅提升,也彰显了中国学术期刊在国际舞台上日益重要的地位。在这样充满变化与活力的学术环境下,接下来将为大家介绍一篇发表于 iMeta 期刊的论文,让我们一同深入探究其蕴含的学术价值 。
https://doi.org/10.1002/imt2.70038

正式介绍
基本信息
-
论文标题:使用单细胞分析平台对单细胞多组学数据进行综合分析
-
发表期刊:iMeta,中科院微生物学大类分区1区Top,IF=33.2000
-
发表日期:2025年4月28日在线发表
研究背景
单细胞组学技术发展使研究能深入到单细胞层面,但现有分析平台存在局限,如多针对单一模态数据,且使用复杂,需要大量编码知识,难以满足当前多组学数据分析需求,尤其对于流行的10× Genomics组学数据类型,缺乏全面且无需编码的解决方案。
研究思路
开发这一基于网络的平台,整合多种先进算法、交互可视化功能和直观界面,支持多种单细胞组学数据类型及空间转录组学分析。通过自动化关键分析步骤、提供丰富分析工具和可视化结果,降低分析门槛,使研究人员无需编程技能即可进行综合分析。
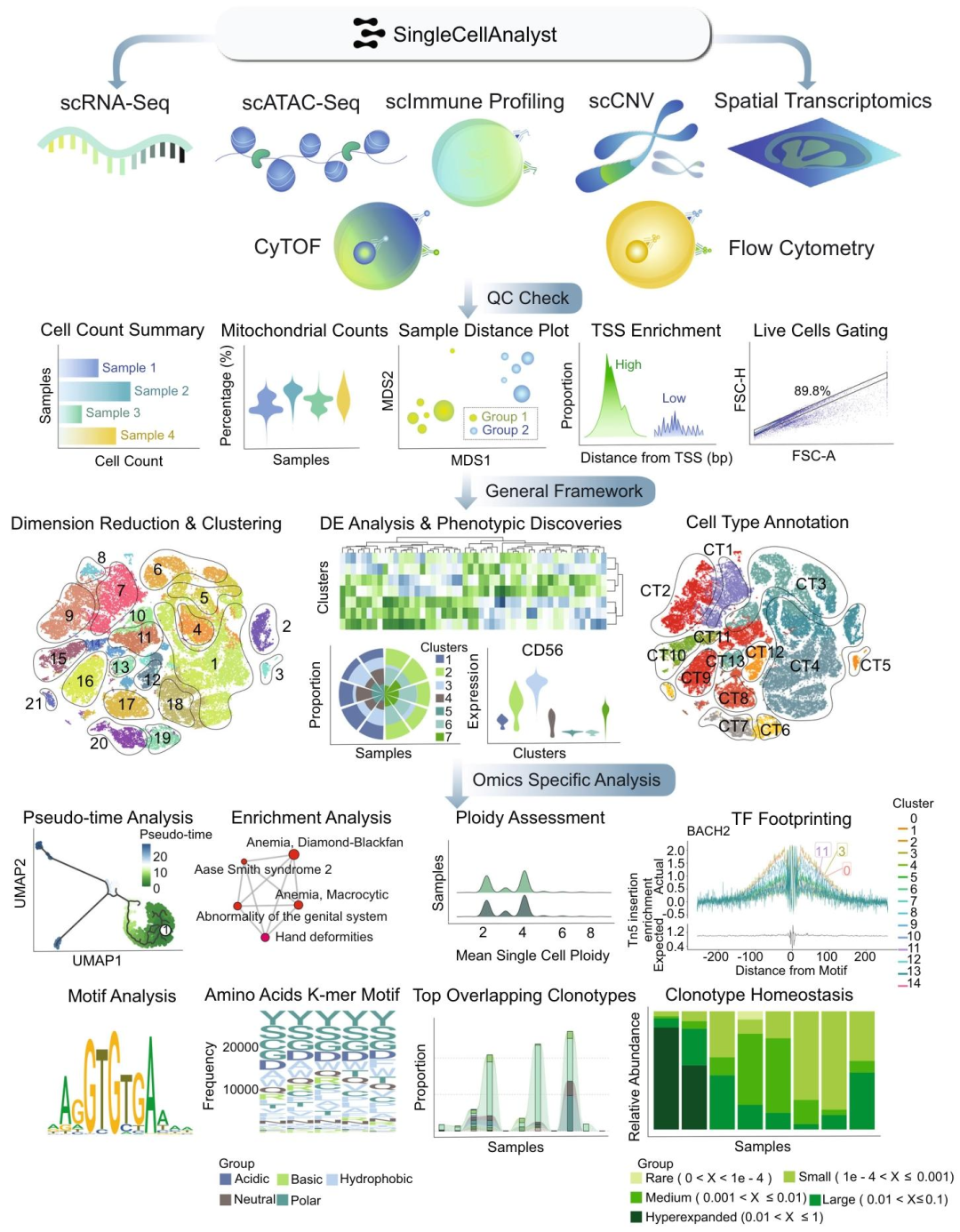
图1:单细胞分析平台网页服务器上的分析框架概述

图2:单细胞分析平台数据库概述
研究亮点
多组学覆盖全面:支持六种单细胞组学类型和空间转录组学,涵盖几乎所有前沿单细胞组学技术,远超其他同类平台。操作简便易用:无需编码,界面直观友好,简化工作流程,降低学习成本,使不同技术背景的研究人员都能轻松使用。功能丰富强大:自动化质量控制、数据处理和表型分析等关键步骤,提供20多种交互式分析工具,可进行综合分析和可视化,且分析速度快,能高效利用计算资源。数据连接广泛:与单细胞图谱数据库紧密连接,包含多种组学数据,为比较分析提供丰富资源。
数据来源和生物信息方法
1、数据来源
包括多种单细胞组学数据,如单细胞RNA测序(scRNA - seq)的基因 - 细胞计数矩阵(.csv或.txt格式,或10X Genomics的.h5格式)、单细胞染色质可及性测序(scATAC - seq)的片段文件(.bed)等,还有空间转录组学的表达矩阵(.h5或.csv)及相关元数据等。
2、生物信息方法
使用R语言(v4.2.0)和shiny应用进行前后端集成,依赖seurat(v4.1.0)、Monocle3(v1.0.0)等多种R包。采用主成分分析(PCA)、t - 分布随机邻域嵌入(t - SNE)、均匀流形近似和投影(UMAP)等算法进行降维;k - 最近邻(k - NN)、共享最近邻(SNN)模块化优化算法进行聚类;Wilcoxon秩和检验、逻辑回归等进行差异表达分析;基因集富集分析(GSEA)等进行通路富集分析。
主要结果
1、scRNA-seq分析结果
完成质量控制、数据处理后,进行细胞类型预测、差异表达分析、通路和富集分析以及伪时间轨迹分析。多样本时可进行综合分析,发现表型差异。例如在研究肿瘤微环境异质性时,可通过聚类识别不同细胞群体,差异表达分析找出相关基因,富集分析揭示功能机制。小结:scRNA - seq分析功能全面,能深入研究基因表达动态和细胞状态转变。

图3:scRNA-seq框架的分析工作流程
2、scATAC-seq分析结果
质量控制包括转录起始位点(TSS)富集等检查,后续进行可及染色质区域识别、基序足迹分析等。多样本时整合分析揭示染色质特征。如研究细胞分化过程中,可通过分析染色质可及性与基因表达关系,探索调控元件作用。小结:scATAC - seq分析有助于深入了解染色质调控机制。

图4:scATAC-seq框架的分析工作流程
3、scImmune Profiling分析结果
进行克隆型丰度等质量控制评估,开展免疫库重叠分析、基因使用分析等。多模态数据时整合scRNA-seq数据,研究免疫反应。如在癌症免疫治疗研究中,可分析克隆型扩张与细胞类型关系。小结:scImmune Profiling分析为免疫相关研究提供详细信息。

图5:scImmune Profiling框架的分析工作流程
4、scCNV分析结果
进行倍性评估等分析,识别细胞亚群的基因组异常。多样本时进行系统发育分析和层次聚类,研究CNV模式。在癌症研究中,可用于检测肿瘤细胞的基因组变化。小结:scCNV分析对研究癌症等疾病的基因组变异有重要意义。

图6:单细胞拷贝数变异(scCNV)框架的分析工作流程
5、CyTOF分析结果
处理原始.fcs文件,进行降维和聚类分析,识别细胞表型,包括标记物表达分析、特征选择等。如研究复杂疾病的免疫细胞异质性时,可通过分析细胞表型差异,了解疾病机制。小结:CyTOF分析有助于深入研究细胞表型特征。
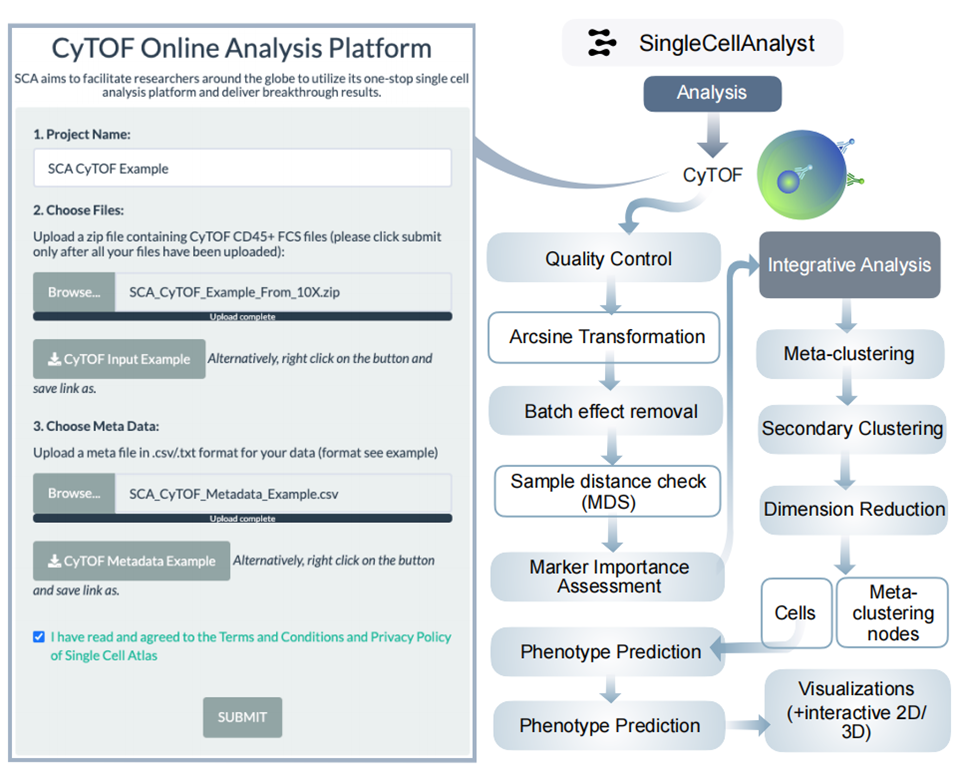
图S4:CyTOF框架的分析工作流程
6、Flow cytometry分析结果
自动化门控策略去除杂质和双峰,进行降维和聚类,评估标记物表达。多样本时综合分析群体差异,可用于免疫监测和生物标志物发现。小结:Flow cytometry分析能精准进行细胞表型分析。

图S5:流式细胞术框架的分析工作流程
7、Spatial transcriptomics分析结果
进行质量控制和数据处理后,进行差异表达分析,将降维和聚类结果可视化在组织图像上。多样本时进行整合分析,结合scRNA - seq数据进行多模态整合。如研究组织发育时,可直观展示基因表达的空间分布。小结:Spatial transcriptomics分析为研究组织空间结构和基因表达关系提供有力工具。
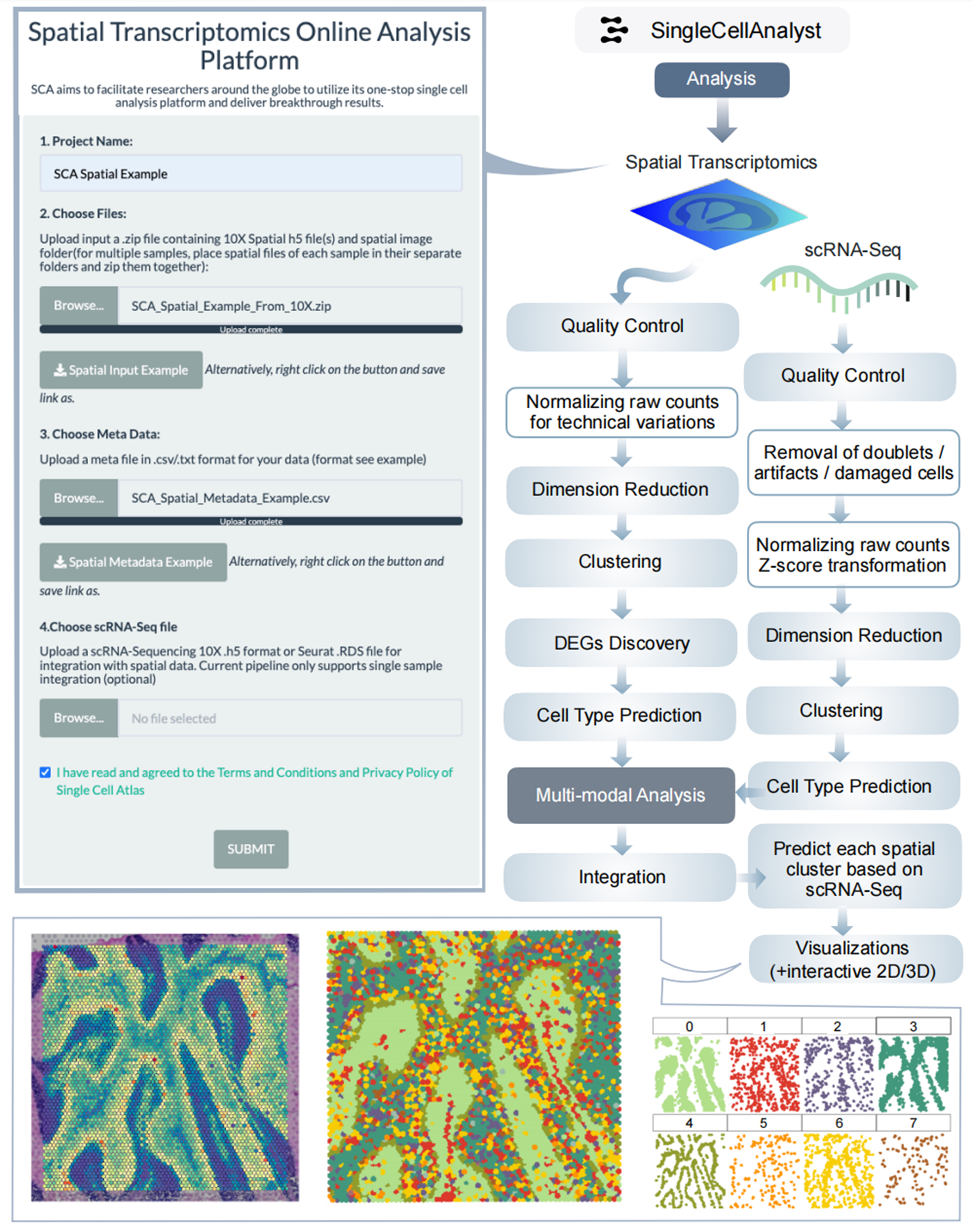
图S3:空间转录组学框架的分析工作流程
研究结论
single-cell analyst支持多种单细胞组学和空间转录组学分析,简化工作流程,自动化关键步骤,提供交互式可视化结果。降低了技术门槛,促进多组学分析,有助于理解生物现象,推动单细胞技术在生物和医学研究中的应用,加速精准医学发展。
研究的局限性和未来方向
目前主要针对人类数据分析,未来计划支持更多物种;加强多模态单细胞数据集联合分析能力,虽docker版本技术上可行,但当前版本为保证一致性暂未实现,后续更新将拓展;增加对更多组学数据类型(如代谢组学)的支持,强化数据整合和预测建模能力,提升计算可扩展性,根据用户反馈持续改进。
感谢您的阅读,欢迎关注“生信学术纵览”。谢谢您的分享、点赞+在看!









)










